

Protein homeostasis refers to the balance of proteins within cells, involving processes such as protein synthesis, folding, modification, transport, function, and degradation. Proteins in cells participate in nearly all life activities, maintaining normal physiological functions. Disruption of protein homeostasis can lead to the occurrence and development of various diseases, including neurodegenerative diseases and cancer. Therefore, maintaining the balance of protein homeostasis is crucial for normal physiological functions in the human body. Traditional targeted drugs mainly block protein function by occupying binding sites, including small molecule inhibitors and monoclonal antibodies. However, due to the complexity of protein function and limitations in targetability, some proteins are difficult to treat using occupancy-driven methods.
Challenges in small molecule targeted drug development
The classical mode of action of small molecule drugs is “occupancy-driven”. Since the FDA approved the first oral small molecule targeted therapy for tumors-tamoxifen targeting the estrogen receptor (ER)-in 1977 for the treatment of breast cancer, small molecule drug development has typically focused on screening for high-affinity inhibitors. Small molecule inhibitors can bind to the active sites or allosteric sites of proteins to abolish their function, which is an “occupancy (of a specific site on the target protein) driven (loss of function of the protein)” mode of action.
Small molecule inhibitors face two major challenges: resistance mutations and difficult-to-drug targets. On the one hand, patients who respond to small molecule inhibitors eventually develop resistance, mainly due to resistant mutations. On the other hand, the “occupancy-driven” mode of action requires explicit pockets/grooves on the target protein that can be occupied by small molecules. However, some protein pockets/grooves are very shallow or even lack catalytic regions, resulting in less than ideal effects of small molecule inhibitors. These target proteins are often referred to as difficult-to-drug targets.
Protein targets that act in the form of protein complexes
Inhibiting one subunit of a protein complex is difficult to completely deactivate the complex. Additionally, protein-protein interactions are difficult to block with small molecules. Therefore, protein complexes have traditionally been considered difficult-to-drug targets, such as the PRC protein complex involved in epigenetic regulation and the BAF chromatin remodeling complex.
Protein targets with scaffold functions
Some proteins do not depend on or lack enzymatic catalytic sites themselves but indirectly exert their function by binding/recruiting other proteins. Binding of small molecule drugs to such proteins is difficult to completely abolish the function of the target protein, with representative proteins such as focal adhesion kinase (FAK). Recently, BTK has also been found to have scaffold functions.
Protein targets with numerous homologous genes
Some proteins have multiple homologous genes, and these homologous proteins have highly similar structures and sequences (especially in kinase active regions) but different functions. Selectivity of small molecule inhibitors for specific target proteins may be relatively limited, leading to significant side effects, with representative proteins such as the CDK family.
Due to limitations of small molecule inhibitors, two-thirds of potential targets have not been drugged. There are approximately 20,000 protein-coding genes in the human body, and current research indicates that about 10% of these proteins are associated with disease, accounting for approximately 2,000 potential targets. However, according to a 2017 review by NRDD, at that time, the FDA had approved only 667 target proteins out of 1,578 drugs, with small molecule drugs targeting 549 and large molecule drugs targeting 146, accounting for only one-third of potential targets. A large number of potential targets are limited by the limitations of small molecule drugs in drug development.
Two Major Systems Based on Protein Degradation
In eukaryotic cells, there are two major systems responsible for protein and organelle degradation: the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system.
The ubiquitin-proteasome system (UPS) is the core mechanism for degrading damaged or misfolded proteins inside cells, participating in the degradation of over 80% of cellular proteins. Its main process involves: first, ubiquitin (Ub) forms a thioester bond with ubiquitin-activating enzyme (E1); activated ubiquitin is transferred to ubiquitin-conjugating enzyme (E2) through thioester exchange reaction; then, ubiquitin ligase (E3) simultaneously binds to the “E2-ubiquitin complex” and the target protein, bringing them into close proximity in space, driving ubiquitin transfer to the target protein. This process is ATP-dependent, and repeated cycles can mark substrates with ubiquitin chains, ultimately recognized and degraded by the proteasome.
In addition to the UPS, another degradation pathway mediated by lysosomes exists widely in eukaryotes. This pathway involves a wide range of substrates, including senescent or damaged organelles, protein aggregates, and invading pathogens, complementing the UPS. The lysosomal degradation pathway mainly includes the endosome-lysosome pathway (ELP) and the autophagy-lysosome pathway (ALP). Therefore, targeted degradation techniques based on ELP are mainly applied to extracellular proteins and membrane proteins, such as LYTAC; ELP refers to the process in which cells fuse extracellular substances or membrane proteins with endosomes through receptor-mediated endocytosis and ultimately transport them to lysosomes for degradation. ALP usually refers to the process of wrapping substrates in autophagosomes with the participation of key autophagy protein LC3 and eventually fusing with lysosomes for degradation. Targeted degradation techniques based on ALP further expand the technical means for targeting intracellular proteins (including protein aggregates) and have been successfully applied to degrade damaged organelles and other non-protein biomolecules.
Based on the two pathways mentioned above, targeted protein degradation technologies operate on an “event-driven” rather than an “occupation-driven” mode of action. In theory, they only require catalytic amounts of drugs to exert strong therapeutic effects, showing significant advantages in targeting “difficult-to-drug” targets, improving selectivity, overcoming resistance, and reducing toxic side effects, with broad application prospects.
PROTAC Products at BOC Sciences
| Products Name | Description |
| E3 Ligase Ligand-Linker Conjugate | E3 ligase ligand-linker conjugate is a range of synthetic compounds containing E3 ligase ligands and connectors used in the PROTAC technology. |
| Ligand for E3 Ligase | There are hundreds of E3 ligases in cells, but only a limited number of them are successfully used in reported PROTACs, such as VHL (von Hippel-Lindau disease tumor suppressor protein), CRBN (Cereblon), MDM2 (the mouse double minute 2 homologue) and IAP (inhibitor of apoptosis). |
| Ligand for Target Protein | The ligand for target protein will result in the attachment of PROTAC to the protein of interest for ubiquitination and subsequent degradation. |
| PROTAC Linker | By far the most common motifs in PROTAC linker are PEG and alkyl chains of various lengths, which are the only motifs in approximately 55% and 30% of connectors respectively. |
| SNIPER | SNIPERs (specific and nongenetic IAP-dependent protein erasers) are a class of small molecule protein degraders consisting of ligands of apoptosis proteins (inhibitor of apoptosis proteins) chimerised with those of target proteins. |
| Molecular Glue | Molecular glue is a kind of small chemical molecules that act on the interface between protein and protein. |
Advantages of PROTAC Technology
Compared to traditional small molecule inhibitors, PROTACs can degrade non-enzymatic proteins, undruggable proteins, etc. They are expected to have higher activity and selectivity and possess a catalytic effect, potentially enabling low-dose, low-frequency dosing.
Compared to monoclonal antibodies, PROTACs can target a large number of intracellular targets, with stronger tissue penetration, and can be administered orally.
Both siRNA and PROTACs can achieve protein degradation, but siRNA has a short half-life and is easily degraded, and drug delivery remains a critical limiting factor for its application.
PROTAC technology has comprehensive advantages in targeting undruggable proteins, overcoming resistance, selectivity, and drug administration routes, with great development potential and broad application prospects.
PROTAC Technology at BOC Sciences
| Technology | Description |
| PROTAC-Antibody Conjugates Design | Degrader-antibody conjugates (DACs) are novel entities that combine a PROTAC payload (degrader) with a monoclonal antibody via some type of chemical linker, which have several potential advantages over PROTAC molecules. |
| Light-Controllable PROTAC Technology Development | The light-controllable PROTAC combines the concept of proteolysis targeting chimeric (PROTAC) and photochemistry that is called photopharmacology, which is an emerging field that uses light to precisely control drug activity in spatial and temporal resolution. |
| RNA-PROTACs Technology Development | RNA-PROTACs are novel chimeric structures that mediate the proteasomal degradation of RNA binding proteins (RBPs). |
| Oligonucleotide-based PROTACs Development | Unlike classical PROTACs, which usually target disease-associated proteins with cytoplasmic structural domains containing binding sites, the development of oligonucleotide-based PROTACs has expanded the range of intracellular target proteins, such as DNA-binding proteins. |
| PROTAC Degradation Technology Development | We provide comprehensive PROTAC degradation technology development services. |
Novel PROTAC Technologies
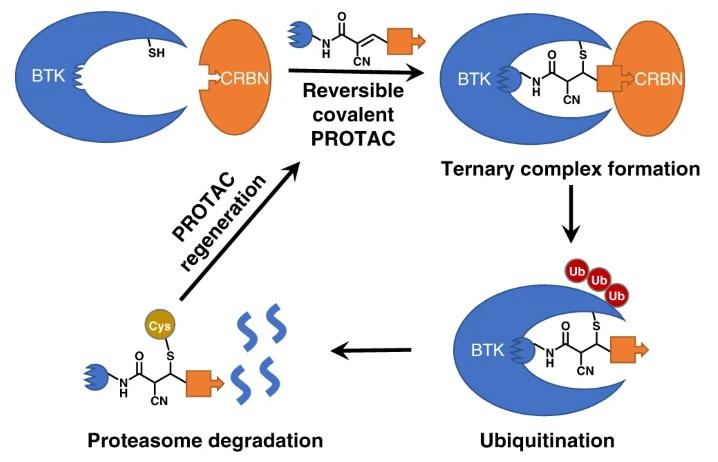
Covalent PROTACs
Currently, the majority of PROTACs bind reversibly to both the target protein and the E3 ligase through reversible interactions. Inspired by covalent small molecule inhibitors, covalent PROTACs have emerged. By introducing electrophilic groups into PROTACs, they can form covalent bonds with the target protein or E3 ligase without compromising their catalytic properties. Therefore, they can maintain longer drug efficacy and show some advantages in improving degradation efficiency and selectivity.
Electrophilic groups capable of forming covalent bonds with the target protein are introduced into PROTACs, such as reversible covalent PROTACs designed based on the electrophilic group cyanoacrylamide.
There are over 600 E3 ligases in the human body, each showing unique substrate specificity. Therefore, designing PROTACs that covalently bind to E3 ligases can enhance PROTACs’ degradation efficiency and selectivity. It is also another approach to develop new E3 ligase ligands.
Homo-PROTAC
Compared to small molecule inhibitors, PROTACs have the advantage of lower effective doses and higher efficiency. However, due to the presence of E3 ligase ligands, PROTACs may interact with other targets besides the target protein, leading to off-target effects and related toxic side effects. For example, CRBN-based PROTACs may not only degrade the target protein but also degrade other targets such as the translation termination factor GSPT1, leading to off-target effects. Homo-PROTAC is a new form of conventional PROTAC, which is directly linked by two identical/different E3 ligase ligands. The dimerization of E3 ligase ligands themselves can dual-hijack E3 ligase to selectively induce their degradation.
The advantage of Homo-PROTAC compared to traditional PROTACs is that they do not bind to other targets, thereby avoiding the occurrence of toxic side effects. Currently, Homo-PROTACs targeting MDM2, VHL, and CRBN have been reported, providing a new method for achieving selective degradation of E3 ligases.
Multi-target PROTAC
Currently, most PROTACs target a single target in the protein family or different subtypes of the same target. Since multi-target inhibitors and bispecific antibodies have been successful, designing PROTACs that can simultaneously degrade two or more different targets may achieve better therapeutic effects and expand the application of PROTAC technology.
Light-controlled PROTAC
PROTACs act through an event-driven mode, and compared to traditional small molecule inhibitors, they can rapidly achieve high activity at low doses in an enzyme-catalyzed manner. However, this makes it difficult to control activity by adjusting drug doses. In addition, similar to small molecule inhibitors, PROTACs may also cause off-target effects.
When administered systemically, PROTACs cause irreversible degradation of both normal cells and tumor cells in the body, leading to the occurrence of side effects. To overcome this problem, researchers have attempted to control protein degradation with high spatiotemporal resolution through external modulation, such as phosphorylation control and light control. Light, due to its convenience, efficiency, and precision, has been widely used as an external control method for regulating protein degradation in recent years.
Light-controlled PROTACs can be divided into two types based on their mechanisms of action: photo-cleavable PROTACs (PC-PROTACs) and photo-switchable PROTACs (PS-PROTACs).
Antibody-PROTAC Conjugates (Ab-PROTAC)
Although numerous PROTACs have been reported targeting different targets, most PROTACs cannot distinguish between different types of cancer cells, leading to poor tissue selectivity. Inspired by antibody-drug conjugates (ADCs), researchers have proposed a novel design strategy called antibody-PROTAC conjugates (Ab-PROTAC) to improve the tissue selectivity of PROTACs.
Ab-PROTAC typically connects an antibody that can specifically target tumor cells with a PROTAC molecule through linkers of different lengths, and theoretically, the linker can be connected to any part of the PROTAC. The hydroxyl group in hydroxyproline on the VHL ligand has been shown to be a suitable connection site. After the PROTAC binds to the antibody, it loses its degradation activity, and is then selectively recognized by tumor cells. The active PROTAC is subsequently released and exerts its degradation activity. Therefore, Ab-PROTACs are expected to exhibit better tissue specificity and lower toxicity.
Folate-PROTAC Conjugates
Folate receptor alpha (FOLR1) is highly expressed in various types of cancer cells and is a common target for targeted drug delivery in anticancer therapy. A series of folate-PROTAC conjugates are obtained by linking folate groups to the hydroxyl group of hydroxyproline on the VHL ligand of PROTACs via ester bonds. The folate moiety can be specifically recognized by FOLR1, and subsequently, FOLR1 mediates the entire conjugate to enter and accumulate in cancer cells. Finally, under the action of endogenous hydrolases in cells, the ester bond in the conjugate is cleaved, releasing active PROTAC to degrade the target protein.
Nucleic Acid Aptamer-PROTAC Conjugates
Nucleic acid aptamers, known as “chemical antibodies,” are single-stranded nucleic acids that can specifically bind to target proteins. Nucleic acid aptamers have advantages such as convenient synthesis, good water solubility, excellent tissue specificity, and penetration. APC 40 is obtained by connecting a highly active BET PROTAC with a nucleolin-targeting aptamer AS1411 through a glutathione (GSH)-sensitive linker. This conjugate can specifically recognize breast cancer cells MCF-7 with high nucleolin expression, and then, in the tumor microenvironment, the linker is cleaved to release the original active PROTAC, which selectively degrades the BRD4 protein. Compared to the original PROTAC molecule, APC 40 has higher tumor tissue selectivity and lower toxicity. It can selectively recognize MCF-7 tumor cells and accumulate in tumor tissues.
As a new generation of drug technology, PROTACs have gradually completed proof-of-concept validation, and some products have achieved positive results in clinical phases I/II. They are expected to overcome resistance to existing products or breakthrough undruggable targets, bringing new opportunities for incremental investment in the small molecule field.