

Antibody conjugated drug (ADC) is a relatively complex molecule that connects a targeted antibody with a cytotoxic payload through a chemical linker. In recent years, people have gradually deepened their research on payload and linker, and made breakthrough progress. ADC represented by Enhertu (DS-8201) breaks through people’s understanding of traditional ADC, and greatly improves clinical therapeutic effect, becoming a blockbuster product. In the past two years, ADC drugs have also ushered in the outbreak of the market, becoming one of the hottest tracks in the field of innovative drugs.
The linker of ADC must not only connect antibodies and toxins, but also need to be able to cleave tumor cells to release toxins. These two functions determine that linker must have two characteristics: First, it must be stable in the body’s circulation system; Second, it must be able to specifically release active ingredients when reaching the tumor cell site. Such a pair of seemingly contradictory requirements makes the development of linker extremely difficult and challenging.

Chemical groups that trigger linker breakage
Cleavable linkers, as the name suggests, are linkers that can be cleaved to release toxins. According to the different cleavage mechanisms, they can be divided into enzymatic cleavage linkers and chemical cleavage linkers.
- Cathepsin cleavable linkers
Common ones are dipeptides (Fig. 2) or tetrapeptide linkers (Fig. 3). These peptides can be cleaved by a variety of cathepsins, and then the cleaved parts undergo a self-elimination reaction to release toxins.


- Phosphatase and pyrophosphatase cleavable linkers
Like cathepsin, phosphatase and pyrophosphatase are also hydrolases that are selectively expressed in lysosomes. To address this feature, a phosphate group or pyrophosphate group can be added to the design of the linker, and then hydrolyzed to release the toxin (Fig. 4).

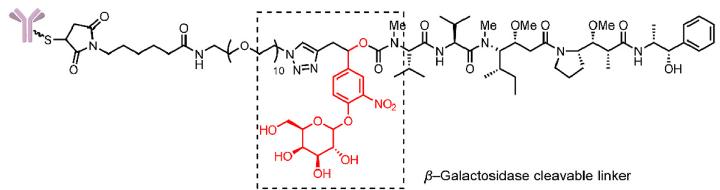
- β-glucuronidase and β-galactosidase cleavable linkers
The linker designed in this design introduces a combination of groups that can be specifically hydrolyzed by these two enzymes and self-degrading spacer. The linker releases toxins through a self-degradation reaction after the β-glucuronic acid and β-galactose groups are hydrolyzed (Fig.5).


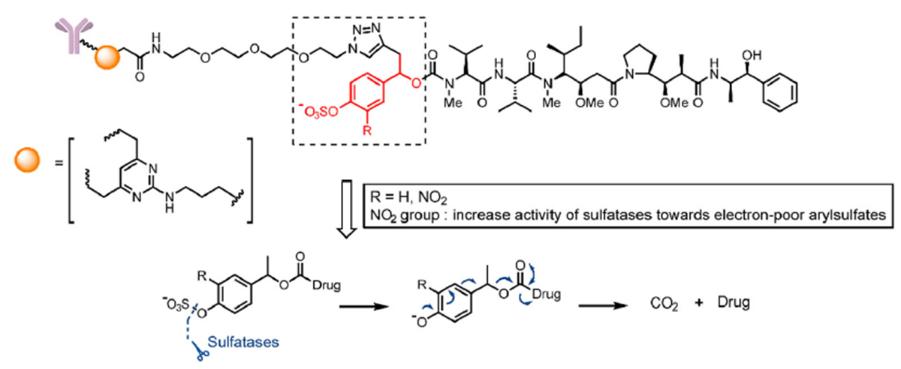
- Sulfonate esterase cleavable linkers
Similar to the design ideas of β-glucuronidase and β-galactosidase, a combination of sulfonate ester group and self-degrading spacer is introduced into the linker. The linker is hydrolyzed by sulfonate esterase and releases toxins through a self-degradation reaction ( Fig. 6).

- Glutathione (GSH)-cleavable linkers
Glutathione lysed linker, that is, linker with disulfide bond groups. The content of glutathione in most tumor cells is higher than that in normal cells, because it has certain reducibility and can reduce disulfide bonds to release toxins (Fig.7).
- Acid–cleavable linkers
Compared with the neutral alkaline pH conditions in normal body fluids and blood, the pH of the tumor microenvironment is generally acidic and can even be as low as 6. This allows the design of linker groups that are sensitive to acidic conditions, such as hydrazone bonds and carbonate esters, which are stable under neutral conditions but can be cleaved under acidic conditions (Fig.8).

New linker technologies are also constantly being developed. Currently, there are several type of linkers that are still in the basic research stage and have not yet been certified by clinical research, such as Fe(II) cleavable linker, light-responsive cleavable linker, and bioorthogonal cleavable linker, etc.
Attachment Sites on the Antibodies for Linkers
The main groups involved in chemical reactions in the antibody molecule are the sulfhydryl group of cysteine and the primary amino group of lysine, which participate in two reactions commonly used in random coupling ADC, namely the Michael addition reaction of sulfhydryl group and maleimide, substitution reaction of amino succinimide ester. In addition, non-natural amino acids can be inserted into the antibody at specific points to introduce reactive carbonyl and azide groups, and linker-drug can be connected by oximation reaction between carbonyl and hydroxylamine and click reaction between azide and alkyne groups. Finally, glutamine is the self-contained antibody, and conjugations can also be achieved by using transglutaminase, which involves enzyme-catalyzed site-directed coupling (Fig. 9).
In addition, another important binding site is the glycosylation site in the antibody structure. The N297 position of the Fc domain of the antibody structure is a conserved glycosylation site. The glycosite-specific ADCs (gsADCs) formed by introducing cytotoxins at this site have the advantages of not affecting antigen binding and having better stability. Advantages have attracted widespread attention in recent years.
Attachment Sites on the Toxin for Linkers
- Carbamate
Carbamate bonds are currently the most commonly used connection method. Payloads containing primary and secondary amine groups can use this connection method. For example, the listed ADC drugs Adcetris and Polivy connect PAB and MMAE through carbamate bond.

- Carbonate Bond
This linker and toxin binding site are both cleavage groups that trigger the release of the toxin. The marketed ADC drug Trodelvy uses this connection method (Fig. 10).

- Amido bond
The amide bond attachment site of Enhertu has a design similar to that of a prodrug. The carboxyl group and amino group of the last glycine of the tetraceptide-linker GGFG are condensed into an amide bond, and the aminomethyl group is self-eliminated after enzymatic digestion, and finally DXD is released (Fig.11).

- Hydrazone bond
It is a releasing group and can also be used as a linker and payload attachment site (Fig.12).

- Disulfide bond
It has the function of connecting toxins. The earliest approved ADC drug Mylotarg and the latest approved ADC drug Elahere use disulfide bonds to connect and release toxins (Fig.13).

References
- Zheng Su, Dian Xiao, Fei Xie, Lianqi Liu, Yanming Wang, Shiyong Fan, Xinbo Zhou, Song Li, Antibody drug conjugates: Recent advances in linker chemistry[J], Acta Pharmaceutica Sinica B 2021;11(12):3889e3907.
- Vesela Kostova, Patrice Désos, Jérôme-Benoît Starck and Andras Kotschy, The Chemistry Behind ADCs[J], Pharmaceuticals, 2021, 14, 442.
- Floris L. van Delft and John M. Lambert, Chemical Linkers in Antibody–Drug Conjugates (ADCs)[M], The Royal Society of Chemistry 2022, Drug Discovery Series No. 81.
- Kyoji Tsuchikama, Zhiqiang An, Antibody-drug conjugates: recent advances in conjugation and linker chemistries[J], Protein & Cell, Volume 9, Issue 1, January 2018, Pages 33–46.
- Wei Shi, Wanzhen Li, Jianxin Zhang, Tiehai Li, Yakai Song, Yue Zeng, Qian Dong, Zeng Lin, Likun Gong, Shuquan Fan, Feng Tang, Wei Huang, One-step synthesis of site-specific antibodyedrug conjugates by reprograming IgG glycoengineering with LacNAc-based substrates[J], Acta Pharmaceutica Sinica B, 2022;12(5):2417-2428.
- Stephen J. Walsh, Jonathan D. Bargh, Friederike M. Dannheim, Abigail R. Hanby, Hikaru Seki, Andrew J. Counsell, Xiaoxu Ou, Elaine Fowler, Nicola Ashman, Yuri Takada, Albert Isidro-Llobet, Jeremy S. Parker, Jason S. Carroll and David R. Spring, Site-selective modification strategies in antibody–drug conjugates[J], Chem. Soc. Rev., 2021,50, 1305.